Polymorphism in 4'-Hydroxyacetophenone: A Molecular Dynamics Simulation Study
Abstract
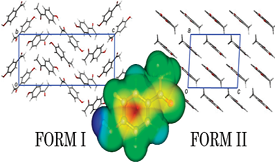
A molecular dynamics simulation study of the two known polymorphs of 4'-hydroxyacetophenone (HAP; form I, monoclinic; form II, orthorhombic) is described. The modeling of the lattice energetics was found to be particularly sensitive to the atomic point charge (APC) selection method, to the number of molecules in the asymmetric unit (Z'), and to the flexibility allowed for the molecules. In order to improve the quality of the APCs, a new strategy that attempts to simulate the polarizability effects of the molecules in the crystal lattice was developed. This method relies in the application of the CHelpG methodology to a molecular aggregate with the same spatial arrangement present in the crystal lattice of the compound. This approach led to ΔtrsHmo(II→I) = +2.4 ± 0.3 kJ/mol and ΔtrsHmo(II→I) = +2.0 ± 0.9 kJ/mol when rigid and flexible models were used, respectively, in good agreement with the corresponding experimental value ΔtrsHmo(II→I) = +0.49 ± 0.13 kJ/mol. Concerning the volumetric properties (density and unit cell parameters), it was concluded that the use of a flexible molecular model was largely insensitive to the chosen methodology for the selection of the APC. Overall, it was concluded that the best performance in the prediction of the energetic and volumetric properties of the two HAP polymorphs was achieved by combining a flexible molecular framework with atomic charges obtained for a molecular aggregate mimicking the crystal packing.
Return Previous Next