Energetics and Structure of Simvastatin
Mol. Pharmaceutics 2013, 10, 2713−2722. DOI: 10.1021/mp400132r
Abstract
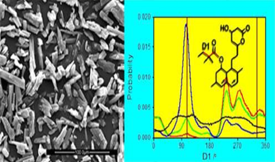
The study of structure−energetics relationshipsfor active pharmaceutical ingredients has received considerable attention in recent years, due to its importance for the effective production and safe use of drugs. In this work the widely prescribed cholesterol-lowering drug simvastatin was investigated by combining experimental (combustion calorimetry and differential scanning calorimetry, DSC) and computational chemistry (quantum chemistry and molecular dynamics calculations) results. The studies addressed the crystalline form stable at ambient temperature (form I) and the liquid and gaseous phases. Heat capacity determinations by DSC showed no evidence of polymorphism between 293 K and the fusion temperature. It was also found that the most stable molecular conformation in the gas phase given by the quantum chemistry calculations (B3LYP-D3/cc-pVTZ) is analogous to that observed in the crystal phase. The molecular dynamics simulations correctly captured the main structural properties of the crystalline phase known from published single crystal X-ray diffraction results (unit cell dimensions and volume). They also suggested that, while preferential conformations are exhibited by the molecule in the solid at 298.15 K, these preferences are essentially blurred upon melting. Finally, the experiments and calculations led to enthalpies of formation of simvastatin at 298.15 K, in the crystalline (form I) ΔfHom (cr I) = −1238.4 ± 5.6 kJ/mol, liquid ΔfHom (l) = −1226.4 ± 5.7 kJ/mol, and gaseous ΔfHom (g) = −1063.0 ± 7.1 kJ/mol states.
Return Previous Next