Evaluation of the OPLS-AA Force Field for the Study of Structural and Energetic Aspects of Molecular Organic Crystals
Abstract
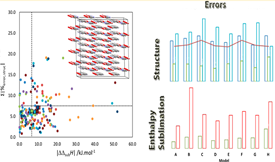
Motivated by the need of reliable experimental data for the assessment of theoretical predictions, this work proposes a dataset of enthalpies of sublimation determined for specific crystalline structures, for the validation of molecular force fields (FF). The selected data were used to explore the ability of the OPLS-AA parameterization to investigate the properties of solid materials in molecular dynamics simulations. Furthermore, several approaches to improve this parameterization were also considered. These modifications consisted in replacing the original FF atomic point charges (APC), by values calculated using quantum chemical methods, and by the implementation of a polarizable FF. The obtained results indicated that, in general, the best agreement between theoretical and experimental data is found when the OPLS-AA force field is used with the original APC or when these are replaced by ChelpG charges, computed at the MP2/aug-cc-pVDZ level of theory, for isolated molecules in the gaseous phase. If a good description of the energetic relations between the polymorphs of a compound is required, then, either the use of polarizable FF or the use of charges determined taking into account the vicinity of the molecules in the crystal (combining the ChelpG and MP2/cc-pVDZ methods) is recommended. Finally, it was concluded that density functional theory methods, like B3LYP or B3PW91, is not advisable for the evaluation of APC of organic compounds, for molecular dynamic simulations. Instead, the MP2 method should be considered.
Return Previous Next